
Bonjour
Je suis bloqué à l'étape de F à G, pourriez vous m'aider
Merci
-----


Bonjour
Je suis bloqué à l'étape de F à G, pourriez vous m'aider
Merci

Je n'ai pas vérifié l'intégralité de ta synthèse mais à mon avis, au lieu d'obtenir le diacide tel que tu le prétends, tu as le diester méthylique (voir 2 étapes avant, MeOH H+ cat.). D'ailleurs mettre un diacide en présence de soude ne fera pas grand grand chose pour l'obtention de ton composé.
Après ce que j'imagine c'est que cette réaction est une condensation de Dieckmann (condensation d'un diester en milieu basique). Cela collerait au niveau du nombre de carbone du composé suivant
Merci
Et vous savez à quoi ça sert ici de faire une Chloration sur le noyau aromatique à l'étape suivante ?
Et je voulais savoir le mécanisme de l'étape de H à I qui est une cyclisation dont on obtient le cycle C
Bonsoir, ton composé F n'est pas le bon.
Ce que tu as dessiné correspond à D.
Les étapes suivantes sont : estérification, puis réduction de la cétone benzylique pour conduire à F
La saponification donne G
On effectue ensuite une chloration. Le groupement méthoxy, donneur, oriente en ortho para. Ici, il va aller en para et protéger cette position pour l'étape suivant qui est une Friedel Crafts. On obtient la bonne formule brute pour I, une cétone comme le suggère l'IR et le cycle C comme le suggère l'énoncé.
Cordialement
Bonsoir,
Je m'y suis collé pour voir, mais que se passe t il à l'étape A ? ^^'
Merci !
Bonjour,
A priori il s'agit d'une condensation de Claisen. Il n'y a qu'un produit formé car un seul des 2 esters est énolisable. On obtient un beta-cétoester, de formule Ph-CO-CH2-CO-OMe
Pourquoi on fait une estérification à l’étape D-->E et pas une protection de la cétone ?
Pour passer de J à K faut il utiliser une base (NaH) en excès ?
1ere déprotonation en alpha de l'acide carbox et attaque sur le diméthyloxalate
2eme déprotonation en alpha de la Cétone
Etape de Cyclisation
3eme déprotonation pour réaliser l'énolate dans le 3eme cycle
Et faire l'énal on protonant avec HOAc
Et enfin décarboxylation avec HCl
??
Pourquoi protéger la cétone vu que ton but c'est de la réduire
Ensuite, tu as fait exactement la même erreur, lors de l'étape de J à K tu as là aussi l'ester méthylique et non l'acide carboxylique. D'ailleurs NaH déprotonerait immédiatement l'acide, donc 1éq serait consommé (et un carboxylate n'est plus du tout réactif, et notamment, plus énolisable)
Ah oui tout à fait j'ai tout corriger ^^
Maintenant je bute sur l'énolate d'alcalins formé à l'étape M
Par contre je ne sais pas ce que ça donne avec le composé électrophile
bonsoir
tu as donc décarboxylé pour arriver à L
tu fais l'énolate de la cétone et le fait réagir sur l'iminium formé entre la diméthylamine et le glyoxylate de tert-butyle
Merci
Avec NaBH4 je résuit la Cétone en alcool
Avec PTSOH je forme une lactone
Mais avec Zn je ne sais pas
Il vient complexer C=O ?
Sachant qu'après il y a H2/Pd/C
Merci Sephiroth ! Maintenant j'ai avancé dans la synthèse et j'ai deux nouvelles questions:
-Lors de la chloration de G a H, on obtient normalement deux composés car le groupe méthoxy oriente en ortho para, donc on devrait une moitié orientée ortho à gauche du méthoxy et une autre avec le chlore en para du och3 ...?
-Je ne vois pas du tout à quoi sert l'acide fluorhydrique, a part son rôle d'acide faible qui déprotonerait les fonctions carboxyles en carboxylates, ni comment on peut cycliser.
Il peut y avoir plusieurs site de chloration mais celui qui est utile dans la synthèse est celle en para du substituant methoxy.
Un acide de DEprotonne JAMAIS un acide ! Au contraire, HF va protonner un fonction carboxyle pour éliminer une molécule d'eau et rendre le carbonyle encore plus électrophile pour le faire réagir selon une réaction de Friedel-Crafts.
l'étape au zinc n'est pas des plus classiquesEnvoyé par Ketur34
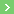
il y a vraisemblablement hydrolyse de la lactone
ensuite, le zinc en milieu acide vient réduire la cétone alpha hydroxylée en cétone (ladite cétone est celle qui est énolisée)
la réduction au palladium vient ôter le chlore
le chlore jouait donc le rôle de groupement protecteur pour orienter la réaction de type Friedel du bon côté.
C'est là qu'en est ProCar
Ketur34 et ProCar font de la synthèse parallèle
@ ZuIIchI : Effectivement, concernant le HF liquide j'ai cru qu'on était dans l'eau donc que F- déprotonerait (éventuellement) l'acide. Mais j'étais un peu loin effectivement. Je continue :gratte: .
Une petite question qui me vient tout à coup:
Est ce que la l'hydrogénation catalytique par le palladium peut hydrogéner des acides carboxyliques ? Autrement je ne vois pas l'intérêt de l'estérification D==>E...
Maintenant je bloque sur J=>K .
NaH est une base forte, donc elle déprotone tous les OH. Egalement, le composé J sous forme énolate réagit avec le dioxalate, et après élimination du méthanolate, puis énolisation (encore une fois), il y a l'intermédiaire que j'ai représenté.
Maintenant à part une attaque nucléophile du O- sur la double liaison, je ne vois pas ce qui peut se passer, étant donné l'impossibilité d'évoluer ensuite...
Merci à tous toujours
Moi j'ai utuilser un deuxième éq de NaH pour deprotoner le H en alpha de la Cétone et d'attaquer sur l'ester pour former un cycle
Moi je bloque à l'étape U, je n'ai aucune idée de ce que peux faire HBr/D sachant qu'on pert C5H10
Avez vous un mécanisme à proposer ?
Comme il n'y a pas de OH à ce stade, je ne vois pas bien comment te répondre.
Le produit J est un tetrahydronaphtalène portant une cétone et un propionate de méthyle. L'hydrure de sodium va déprotonner en alpha de la cétone. L'anion va se condenser sur l'oxalate de diméthyle. On obtient ainsi un diester intermédiaire qui va subir une cyclisation de Dieckmann.
Cordialement
HBr va faire deux choses : déalkylation de l'éther méthylique et hydrolyse du tert-butyle de l'amide
Dans les deux cas, ça commence par une protonation de l'oxygène.
Pour l'éther, Br- vient attaquer le méthyle et libère le OH
Pour l'amide, élimination d'isobutylène
Cordialement
Merci beaucoup
Merci HarleyApril,
Donc hier j'avais fait une image avec chemdraw que j'ai bien sur oublié de poster. Mais pour être tout à fait sur j'ai d'abord représenté mon composé I.
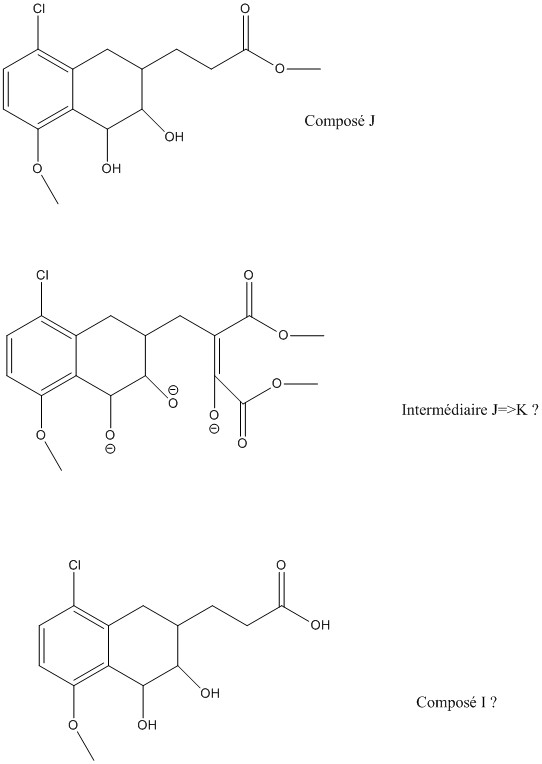
Merci HarleyApril !
Pour en avoir le coeur net j'i représenté I et les suivants:
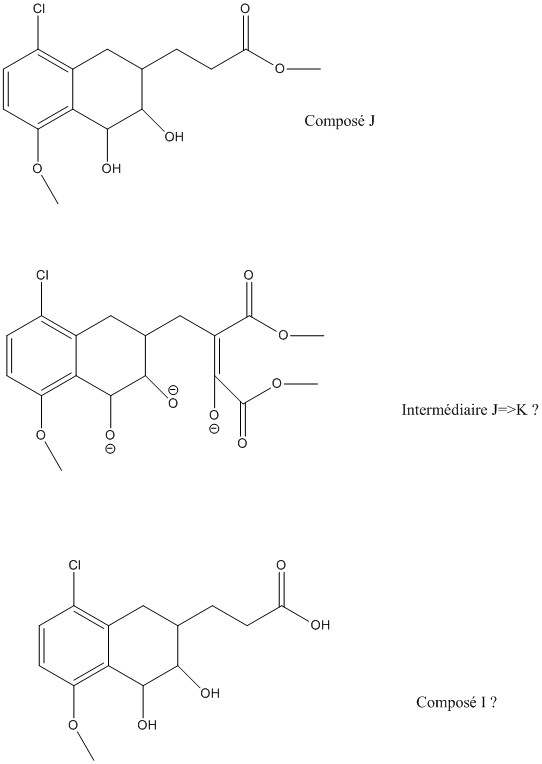
Par contre y a un doublon là, je ne voyais pas mon précédent message, c'est pour ca que j'ai reposté.
c'est bien dommage, mais je persiste à dire qu'il n'y a pas d'alcool et que le produit J est un tetrahydronaphtalène portant une cétone et un propionate de méthyle (cf. post 19)
Comment se passe le mécanisme de la toute dernière étape ?
Selon Tetrahedron 2009 p 10941, le cérium se ferait chélater par les deux oxygènes des carbonyles (cf complexes acac).
Il y aurait ensuite oxydation en radical.
Ledit radical s'additionnerait sur le dioxygène, deux fois, pour conduire à un peroxyde symétrique.
Le peroxyde est ensuite réduit en alcool.
Ils ont montré avec un marquage isotopique que l'oxygène de l'alcool provenait du dioxygène.
Cordialement
